

Function
WiseScaffolder is a stand-alone semi-automatic application for genome scaffolding of pre-assembled contigs using mate-pair data. It also produces editable scaffold maps, allowing either to build gapped scaffolds or usable as a common thread for the manual improvement of scaffolds.
Back to topDescription
WiseScaffolder includes 4 subcommands:
- dumpconfig generates a configuration file that notably specifies the average insert size of the mate-pair library
- preprocess allows the detection and correction of chimerae, the estimation of contigs copy number and produces valuable outputs for the manual improvement of scaffolds
- scaffold constitutes the central scaffold-builder and comprises two modules: i) the interative_scaffold_extender, which works with big, unambiguous contigs, or when they run out, single copy contigs, and ii) the small_contig_inserter, which inserts the small contigs within scaffolds
- buildfasta converts the scaffold(s) map(s) into Fasta sequences.
Classification
Category:
NGS > Scaffolding
User Interface:
Command line, GALAXY wrapper
Operating system:
Any (Python application)
Back to topUsage
The four abovementioned subcommands may be used sequentially as follows:
wisca.py (-p) (-d) (-h) dumpconfig --configout “wisca.conf” (-i 5000) (-b 5000)
→Output : An editable “wisca.conf” configuration file
wisca.py (-p) (-d) (-h) preprocess --configin “wisca.conf” -c “contigs.info” -m “reads_mapping.sam” (--dumpfiles)
→Outputs : chimerae resolution file “chimera.csv”, contig coverage/copy number file “coverage.csv”, additional files dedicated to chimera resolution and manual scaffolding
wisca.py (-p) (-d) (-h) scaffold --configin “wisca.conf” -c “contigs.info”-m “reads_mapping.sam” --scaffoldout “scaffolds_maps.txt” (-k “chimera.csv”) (-v “coverage.csv”)
→Output : An editable “scaffold_maps.txt” file
wisca.py (-p) (-d) (-h) buildfasta -f “contigs.fasta” –s “scaffolds_map.txt”-r “wisca_scaffolds” (-k chimera.csv)
→Output : A “wisca_scaffold” folder containing Fasta-formatted scaffolds
Back to topCommand line arguments
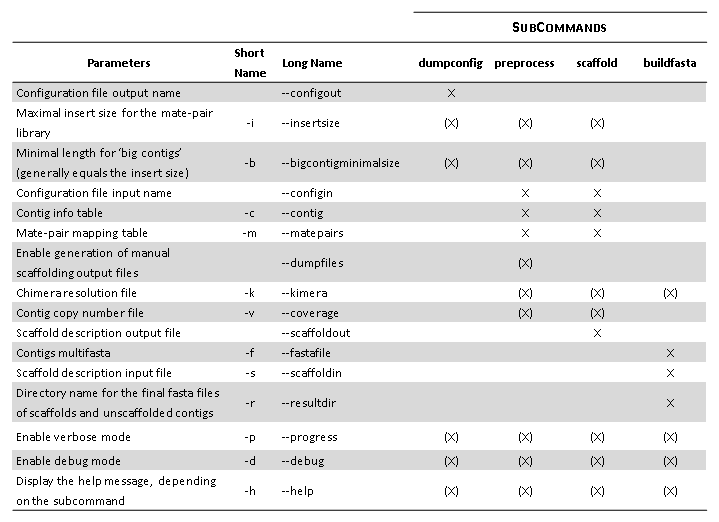
X: parameter required to run a given subcommand
(X): optional parameter. In the case of “insertsize” and “bigcontigminimalsize”, it will take priority over the corresponding parameter in the configuration file.
Input file format
WiseScaffolder requires three inputs:
- Contig info file : tabulated file specifying identifiers, coverage and length of contigs
- Mate-pair mapping file either in SAM format or custom tabulated file
- Multifasta of contigs
Outputs
WiseScaffolder produces the following outputs:
- Configuration file
- Chimerae resolution file
- Contig coverage/copy number file
- Outputs for manual scaffolding
- Mate-pair insert size graph: showing the distribution of the mate-pair insert sizes, as determined using mate-pair reads mapping on the same contig
- Global link map: a symmetric matrix providing for each contig the amount of mate-pair reads linking it to other contigs
- Neighborhood link map: similar to the global link map but with an indication of the location of mate-pair reads on the contig (5' or 3' ends) and their orientation with regard to the contig
- Linkage location map : a symmetric matrix providing for a given contig the average location of MPs linking it to each other contig
- Scaffold maps
- Scaffold fasta
Downloads
Application & Handbook wisca.py v1.0b9 (30 Ko) handbook v1.1 (1 Mo) GALAXY wrapper wrapper v1.0 (5.8 Ko) Test dataset: Synechococcus sp. WH8103 assembly and subset of the mate-pair mapping data WH8103_500x_contigs.fasta (2.3 Mo) WH8103_contigs.info (991 o) | Complementary scripts contigs_renamer.py (2.6 Ko) contig_info_builder.py (4.4 Ko) sam_subsampler.py (4.3 Ko) Python & BioPython |
Reference
Authors
Marine Phototrophic Prokaryotes (MaPP) Team (CNRS-UPMC - UMR7144): Gregory K. Farrant, Frédéric Partensky, Laurence Garczarek
ABiMS Platform (CNRS-UPMC - FR2424): Mark Hoebeke, Gwendoline Andres, Erwan Corre
Please cite
Farrant, G.K., Hoebeke, M., Partensky, F., Andres, G., Corre, E. and Garczarek L., 2015. WiseScaffolder: an algorithm for the semi-automatic scaffolding of Next Generation Sequencing data, in revision for BMC Bioinformatics.
Back to top